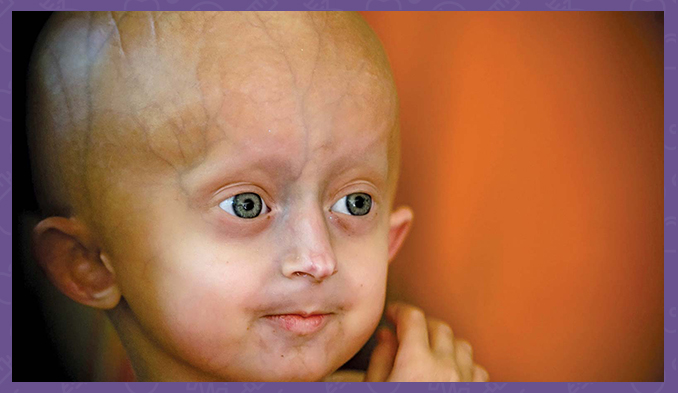
Progeria ya da diğer adıyla Hutchinson-Gilford sendromu en nadir görülen 10 hastalıktan biridir.
Hakkında pek bir şey bilmediğimiz tuhaf bedenler.
Progeria en nadir görülen genetik hastalıklardan biridir. Dünya genelinde yaklaşık 100 kayıtlı hastalık vakası bulunmaktadır. Vücudun aşırı hızlı yaşlanması ile karakterizedir.
Hutchinson-Gilford sendromu, erken çocukluk döneminde dramatik ve hızlı yaşlanma başlangıcı ile karakterize genetik bir durumdur. Etkilenen çocuklar genellikle doğumda ve bebeklik döneminde normal görünürler, ancak daha sonra diğer çocuklardan daha yavaş büyürler ve beklenen oranda kilo almazlar. Şişkin gözler, sivri uçlu ince bir burun, ince dudaklar, küçük bir çene ve çıkıntılı kulaklar gibi karakteristik bir yüz görünümü geliştirirler. Progeria veya Hutchinson-Gilford sendromu ayrıca saç dökülmesine (alopesi), cildin yaşlanmasına, eklem anormalliklerine ve cilt altında yağ kaybına (subkutan yağ) neden olur. Bu durum zihinsel gelişimi veya oturma, ayakta durma ve yürüme gibi motor becerilerin gelişimini etkilemez.
Özellik
Progeria'lı kişilerde erken çocukluk döneminde arterlerde ciddi sertleşme (arterioskleroz) görülür. Bu durum, genç yaşta kalp krizi veya felç geçirme olasılığını büyük ölçüde artırır. Bu ciddi komplikasyonlar zamanla daha da kötüleşebilir ve etkilenen kişiler için hayati tehlike oluşturabilir. Bu durum çok nadir görülür. Dünya genelinde her 4 milyon yenidoğandan 1'inde görüldüğü bildirilmektedir.
Hutchinson-Gilford sendromu/progeria otozomal dominant bir durum olarak kabul edilir, yani her bir hücrede değişmiş genin bir kopyası bozukluğa neden olmak için yeterlidir. Bu durum LMNA genindeki yeni mutasyonlardan kaynaklanır ve neredeyse her zaman ailesinde böyle bir bozukluk öyküsü olmayan kişilerde görülür.
Progeria genellikle aile içinde bulaşmaz. Gen değişikliği neredeyse her zaman rastgele bir olaydır ve son derece nadirdir. HGPS/ Hutchinson-Gilford sendromu/ olmayan diğer progeroid sendrom türlerine sahip çocuklarda aileden geçen hastalıklar olabilir. Ancak HGPS, sporadik otozomal dominant mutasyondan kaynaklanmaktadır. Sporadiktir çünkü o ailede yeni bir değişikliktir ve baskındır çünkü sendroma sahip olmak için genin yalnızca bir kopyasının değişmesi gerekir. Hiç progeria hastası çocuğu olmamış ebeveynler için progeria hastası bir çocuk sahibi olma ihtimali 4-8 milyonda 1'dir, ancak daha önce progeria hastası bir çocuğu olanlar için bu ihtimal yaklaşık 2-3%'ye yükselmektedir. Bunun nedeni, ebeveynlerden birinin hücrelerinin küçük bir kısmında progeria için genetik mutasyon bulunması ancak progeria olmaması şeklindeki mozaiklik adı verilen bir durumdur.
Teşhis
Progeria genellikle bir çocuğun yaşamının ikinci yılında veya daha sonra, progeroid özellikler fark edilmeye başladığında teşhis edilir. Tanı, kapsamlı bir klinik değerlendirme, karakteristik fiziksel bulgular, dikkatli bir hasta öyküsü ve tanısal genetik testlere dayanır. Daha az yaygın olarak, bazı şüpheli belirtilerin tanınmasına dayanarak doğumda sendromun varlığından şüphelenilebilir.
Uygun bir öykü için, hastalıkla potansiyel olarak ilişkili belirli iskelet anormalliklerini doğrulamak veya karakterize etmek için özel görüntüleme testleri yapılabilir, örneğin
- bazı parmak kemiklerinde (terminal falankslar) dejeneratif değişiklikler (osteoliz)
- ve/veya kalça yuvası (asetabulum).
Tanı, ilişkili kardiyovasküler anormallikleri değerlendirmek ve uygun hastalık yönetimini belirlemek için kapsamlı kardiyak değerlendirmeler ve sürekli izleme (örn. klinik muayeneler, röntgenler, özel kardiyak testler) ile tamamlanır.
Terapi türleri. Tedavi
Eylül 2012'de, bir çocukluk çağı progeria ilacı için yapılan ilk klinik çalışmanın sonuçları, başlangıçta kanser tedavisi için geliştirilen bir tür farnesiltransferaz inhibitörü (FTI) olan Lonafarnib'in progeria için etkili olduğunu ortaya koymuştur. İlacı alan çocuklar dört temel parametreden bir veya daha fazlasında iyileşme göstermiştir:
- ekstra kilo alımı
- daha iyi işitme
- gelişmiş kemik yapısı
- ve en önemlisi - kan damarlarının esnekliğinin artması
FDA onaylı olmayan ve bu nedenle yalnızca klinik ilaç denemeleri yoluyla elde edilebilen lonafarnib dışında, HGPS (Hutchinson-Gilford progeria sendromu) etkilenen her bireyde görülen spesifik semptomları hedefler. Hastalığın yönetimi, etkilenen çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilecek profesyonellerden oluşan bir ekibin koordineli çabalarını gerektirebilir. Ekip şunları içerebilir:
- çocuk doktorları
- iskelet, kaslar, eklemler ve diğer ilgili dokuların bozukluklarını teşhis ve tedavi eden doktorlar (ortopedistler)
- kalp ve ana kan damarlarındaki anormallikleri teşhis ve tedavi eden doktorlar
- fizyoterapistler
Daha fazla bilgi için Medical Karaj olarak hizmetinizdeyiz.
Bizi aşağıdaki numaralardan arayın "Tıbbi Karaj": 0879 977 401 veya 0879 977 402.
Ayrıca sürekli güncellenen Facebook içeriğimize de göz atın.