La progeria, nota anche come sindrome di Hutchinson-Gilford, è una delle 10 malattie più rare.
Gli strani corpi di cui non sappiamo molto.
La progeria è una delle malattie genetiche più rare. Sono circa 100 i casi registrati di questa malattia in tutto il mondo. È caratterizzata da un invecchiamento estremamente rapido dell'organismo.
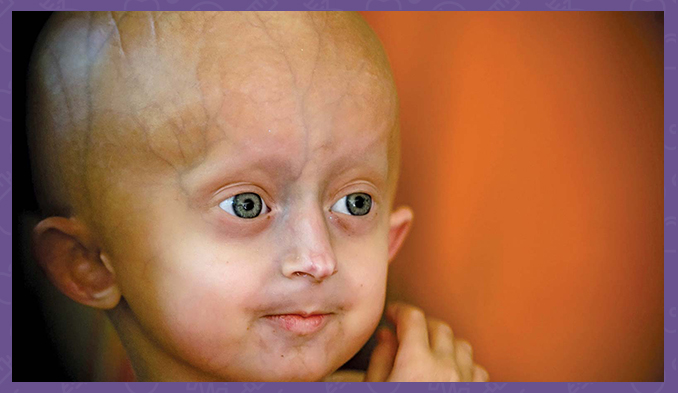
La sindrome di Hutchinson-Gilford è una condizione genetica caratterizzata dall'insorgenza rapida e drammatica dell'invecchiamento nella prima infanzia. I bambini affetti da questa patologia appaiono di solito normali alla nascita e durante l'infanzia, ma crescono più lentamente degli altri e non aumentano di peso al ritmo previsto. Sviluppano un aspetto facciale caratteristico, che comprende occhi sporgenti, naso sottile con punta appuntita, labbra sottili, mento piccolo e orecchie sporgenti. La progeria, o sindrome di Hutchinson-Gilford, causa anche perdita di capelli (alopecia), invecchiamento della pelle, anomalie articolari e perdita di grasso sotto la pelle (grasso sottocutaneo). Questa condizione non influisce sullo sviluppo intellettuale o sullo sviluppo delle capacità motorie, come stare seduti, in piedi e camminare.
Caratteristica
Le persone affette da Progeria presentano un grave indurimento delle arterie (arteriosclerosi) nella prima infanzia. Questa condizione aumenta notevolmente le probabilità di avere un infarto o un ictus in giovane età. Queste gravi complicazioni possono aggravarsi nel tempo e mettere a rischio la vita delle persone colpite. La condizione è molto rara. È stato riferito che si verifica in 1 su 4 milioni di neonati in tutto il mondo.
La sindrome di Hutchinson-Gilford/progeria è considerata una condizione autosomica dominante, il che significa che una copia del gene alterato in ogni cellula è sufficiente a causare il disturbo. La condizione deriva da nuove mutazioni nel gene LMNA e si verifica quasi sempre in persone che non hanno una storia familiare di tale disturbo.
La progeria di solito non si trasmette in famiglia. L'alterazione genica è quasi sempre un evento casuale, estremamente raro. I bambini affetti da altri tipi di sindromi progeroidi che non sono la HGPS/sindrome di Hutchinson-Gilford possono avere malattie che si trasmettono in famiglia. La HGPS, tuttavia, è dovuta a una mutazione sporadica autosomica dominante. Sporadica perché si tratta di un cambiamento nuovo in quella famiglia e dominante perché basta una sola copia del gene per avere la sindrome. Per i genitori che non hanno mai avuto un figlio con progeria, le probabilità di avere un figlio con progeria sono di 1 su 4 - 8 milioni, ma per coloro che hanno già avuto un figlio con progeria, le probabilità aumentano a circa 2-3%. Ciò è dovuto a una condizione chiamata mosaicismo, in cui un genitore presenta una mutazione genetica per la progeria in una piccola parte delle sue cellule, ma non presenta progeria.
Diagnosi
La progeria viene solitamente diagnosticata nel secondo anno di vita del bambino o più tardi, quando le caratteristiche progeroidi iniziano a diventare evidenti. La diagnosi si basa su un'accurata valutazione clinica, su reperti fisici caratteristici, su un'attenta anamnesi e su test genetici diagnostici. Meno comunemente, la presenza della sindrome può essere sospettata alla nascita, sulla base del riconoscimento di alcune manifestazioni sospette.
Per un'anamnesi corretta, possono essere eseguiti esami di diagnostica per immagini specializzati per confermare o caratterizzare alcune anomalie scheletriche potenzialmente associate alla malattia, come ad esempio:
- alterazioni degenerative (osteolisi) di alcune ossa delle dita (falangi terminali)
- e/o della cavità dell'anca (acetabolo).
La diagnosi è completata da valutazioni cardiache approfondite e da un monitoraggio continuo (per esempio, esami clinici, radiografie, test cardiaci specializzati) per valutare le anomalie cardiovascolari associate e determinare la gestione appropriata della malattia.
Tipi di terapia. Trattamento
Nel settembre 2012, i risultati del primo studio clinico in assoluto per un farmaco contro la progeria infantile hanno rivelato che Lonafarnib, un tipo di inibitore della farnesiltransferasi (FTI) originariamente sviluppato per il trattamento del cancro, si è dimostrato efficace per la progeria. I bambini che hanno assunto il farmaco hanno mostrato miglioramenti in uno o più di uno dei quattro parametri chiave:
- aumento di peso extra
- un udito migliore
- miglioramento della struttura ossea
- e, soprattutto, una maggiore flessibilità dei vasi sanguigni.
A parte il lonafarnib, che non è approvato dalla FDA e quindi è disponibile solo attraverso studi clinici, il trattamento della HGPS (progeria Hutchinson-Gilford sindrome) si rivolge ai sintomi specifici che si manifestano in ogni individuo colpito. La gestione della malattia può richiedere gli sforzi coordinati di un team di professionisti che possono avere bisogno di pianificare in modo sistematico e completo il trattamento del bambino affetto. Il team può comprendere:
- pediatri
- medici che diagnosticano e trattano i disturbi dello scheletro, dei muscoli, delle articolazioni e di altri tessuti connessi (ortopedici)
- medici che diagnosticano e trattano le anomalie del cuore e dei suoi principali vasi sanguigni
- fisioterapisti
Per ulteriori informazioni, noi di Medical Karaj siamo a vostra disposizione.
Chiamateci ai seguenti numeri "Karaj medico": 0879 977 401 o 0879 977 402.
Tenete d'occhio anche i nostri contenuti costantemente aggiornati su Facebook.